|
A centralized state-of-the-art proteomics laboratory has been
established at K-State for the detailed structural analysis of
proteins/peptides by mass spectrometry. Facility is equipped a 2-D PAGE
apparatus supported by robotic workstations that image, excise, digest, and
stage protein samples for MALDI-TOF or MALDI-TOF/TOF analyses. The core
facility will house an MS instrument, which will have the capability to
generate peptide mass fingerprint (PMF) data on proteolytic digests and
reference peptide signatures against spectral libraries for identification. If
identifications are inconclusive, the MS instrument has the capability to
isolate specific peptide precursor ions for further dissociation and generation
of amino acid sequence information for characterizing structure and
post-translational modifications (PTM’s).
In addition to implementing
the above 2d-PAGE (gel-based) strategy, two additional sample preparation
workstations are available that utilize a new proteomics strategy of separating
digested protein mixtures by nano-flow liquid chromatography, followed by a
post-column 1:1 split of effluent for analysis by ESI and MALDI - one half of
the split effluent is delivered directly on-line to an ESI-Ion Trap MS/MS
system - the other half is fraction collected onto a MALDI sample target at
timed intervals for MALDI-TOF and/or MALDI-TOF/TOF analysis (typically known as
LC-MALDI). 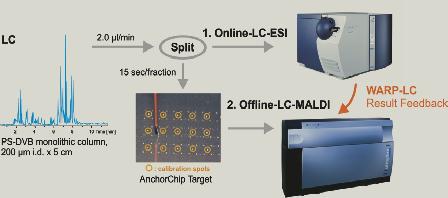 The benefit of this new strategy is threefold: 1) the LC separation
prior to MALDI analysis eliminates the ion-suppression/charge competition
effects typically seen with peptide mixtures and, as a result, leads to a
greater number of positively identified proteins; 2) the MALDI target with
time-collected fractions becomes a “snapshot” in time of the LC run and, by
having no temporal constraints, can be re-analyzed at any time using different
experimental parameters to mine the data for additional proteomic content; 3)
by obtaining both ESI-MS/MS data and MALDI-TOF/TOF data in a single nano-flow
LC run, complementary sequence information is obtained due to different MS/MS
dissociation mechanisms. Combining ESI and MALDI data as merged files increases
amino acid residue coverage over what is typically seen with ESI-MS/MS or
MALDI-TOF/TOF separately. This combined capability greatly enhances the ability
to identify proteins by library search or through de novo sequencing methods. The benefit of this new strategy is threefold: 1) the LC separation
prior to MALDI analysis eliminates the ion-suppression/charge competition
effects typically seen with peptide mixtures and, as a result, leads to a
greater number of positively identified proteins; 2) the MALDI target with
time-collected fractions becomes a “snapshot” in time of the LC run and, by
having no temporal constraints, can be re-analyzed at any time using different
experimental parameters to mine the data for additional proteomic content; 3)
by obtaining both ESI-MS/MS data and MALDI-TOF/TOF data in a single nano-flow
LC run, complementary sequence information is obtained due to different MS/MS
dissociation mechanisms. Combining ESI and MALDI data as merged files increases
amino acid residue coverage over what is typically seen with ESI-MS/MS or
MALDI-TOF/TOF separately. This combined capability greatly enhances the ability
to identify proteins by library search or through de novo sequencing methods.
Another important feature of the MALDI TOF/TOF instrument is its
ability to be used for MALDI-Imaging of specific proteins in tissues. This new technique can be used to identify
specific proteins with mass ranges of 500-70,000 Da.
Workflow Management and Data
Interpretation: The Bruker system that
we have purchased allows for complete software intergration for all of the
system components, including sample tracking through transponder reading in the
MALDI targets. There are also a
comprehensive set of software tools for
validation and reporting of results. There are separate programs for the quick
evaluation of entire LC-MS/MS runs; a protein browser program to identify
peptides and proteins; BioTools TM software for automated and
interactive interpretation of MS and MS/MS data that help identify mutations,
polymophisms; post-translational or chemical modifications; and de novo sequencing. There is also a
program called ProteinScape TM that provides database solutions for
proteomics projects. This program will send and retrieve data from our in-house
server that has the mass spectral protein data base-- Mascot TM. Print outs can be generated that label 2d-gel
pictures with the proteins’ identities for the various spots.

|

